细菌16S rRNA测序能否鉴定到属种(species),为什么保藏中心将16S测序作为鉴定依据?
来源:武汉市灰藻生物科技有限公司 浏览量:1369 发布时间:2025-11-14 20:54:46
引言
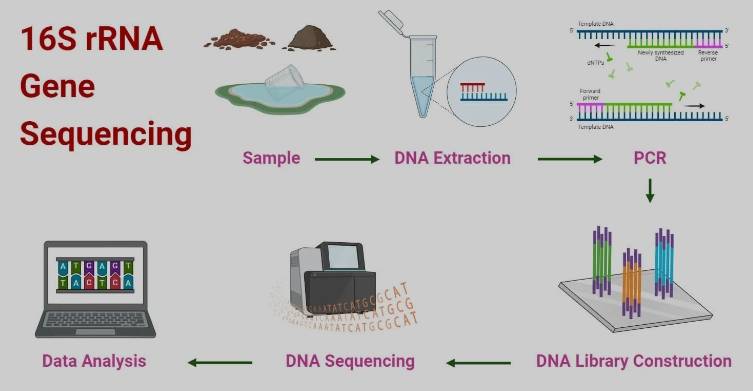
基于扩增子的细菌(包含古细菌)16S rRNA基因测序技术,是目前最经济高效的初步鉴定工具,尤其适合大规模菌株初筛,通常也是保藏中心鉴定细菌身份的“首选”手段
保藏中心确认细菌身份,为什么如此信赖16S测序?
16S rRNA测序技术是否能精确区分细菌的属种,甚至具体株型?
本次我们将围绕这些问题,结合专栏《细菌和古菌16S rRNA基因测序:原理、步骤、用途与示意图》(访问链接:https://www.huizaobio.com/article/a390.html),对16S rRNA测序技术进行更深入的挖掘和解析。
16S rRNA测序
为什么保藏中心依赖16S测序技术确认细菌身份?
首先,需明确的是,菌种保藏是一项偏基础科研建设的工作,通常由政府出资或牵头融资筹建,从立项起便将是一份旷日持久的系统工程。
关于保藏中心的介绍,另一专栏《微生物资源保藏中心的成立背景、目标使命、质量标准和其它相关事项说明(基于世界微生物联合委员WFCC指南)》(访问链接:https://www.huizaobio.com/article/a365.html),我们已经做了详尽介绍,不在此过多赘述。
基于保藏中心的定位,我们应当知道,菌种保藏工作耗时耗力,对稳定性要求较高,因此每一项技术的投入使用,均须斟酌。
16S rRNA基因测序,作为保藏中心最常用的,新分离培养物的初筛手段,正是考虑其在鉴定上,表现出的高效性、经济性和稳定性,同时结合不断成熟的模式株参照数据库,也符合保藏中心标准化流程的体系构建。
将16S序列与数据库中有效发表的模式株(validly published type strains),进行BLAST或系统发育树分析,根据相似度,将其作为新种候选或暂定种名。
事实上,鉴定工作通常已由原始分离人完成,保藏中心进一步审核确认。当然,随着新数据出现(如全基因组发布),原有基于基于16S的分类,是可能被修正的,这部分工作通常由保藏中心完成。
菌种纯培养物的DNA来源单一,PCR扩增目标明确特异性强,不易产生非特性性条带,测序准确性非常高,因此一代16S(Sanger测序)仍是保藏中心和微生物实验室的鉴定“金标准”。
综上,保藏中心选择16S鉴定细菌,是因为16S测序的价格便宜、使用便利,并且有庞大的模式细菌文库作为参照,保证准确性。
16S rRNA基因测序,能否鉴定到具体的细菌种属?
对于这个问题,首先要区分来源是,混合样本(如粪便,土壤,污水等),还是单菌纯培养物(挑单或稀释纯化),不同样本需解决的问题略有差别。
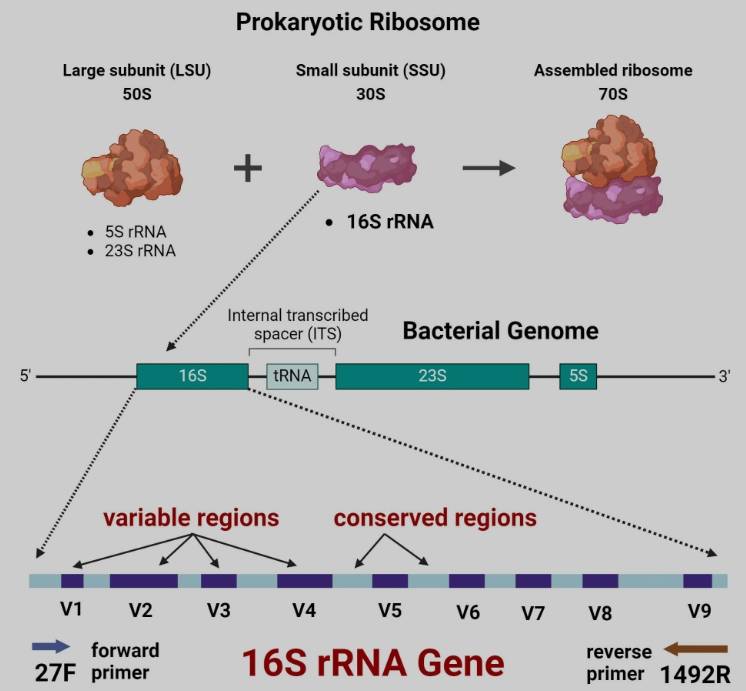
16S测序针对的是,原核生物30S小亚基中的一小段16S核糖体RNA(rRNA),基因全长约1500个碱基对(bp)。
16S序列结构,兼具保守区和可变区,其中可变区决定了种分类,整个16S rRNA包含9个高变区V1-V9。
对于混合样本,二代测序只检测部分可变区,很难鉴定到属。随着高通量测序技术的普及,针对整个V1-V9全长区域测序,分辨率更高,更容易鉴定到种水平。
目前,16S全长测序使用更频繁,不过混合样本的16S信号混杂,需通过聚类算法降噪区分物种,且对于低丰度污染难识别。
对于纯培养物,16S测序的DNA单一,通用引物可扩增出相对纯净的16S序列,无需聚类可合并出高质量的单一识别序列。
整个序列直接比对数据库(EZBioCloud/NCBI等),获取最相近的模式株,确认种级分类。
也就是说,16S不能在所有情况下,稳定鉴定到种;但在纯培养、数据库完善、种间差异明确的前提下,完全可以鉴定到种。
同时,应当认识到,16S只是分类工作的起点,宜配合其它手段,复核鉴定结果。
16S rRNA测序
对于许多关系密切的物种,16S序列高度相似,如何区分?
许多亲缘关系密切的物种(如 Bacillus cereus、B. anthracis 和 B. thuringiensis,16S序列高度相似(>99%),难以准确区分。
可结合不同基因水平的检测手段,对其深入鉴定:
宏观表征水平:表型特征分析,如形态、生化、生理特性等;
基因片段水平:除16S之外的看家基因,如rpoB(RNA polymerase β-subunit gene), gyrB(DNA gyrase subunit B gene), recA(Recombinase A gene), dnaK(Heat shock protein 70 (Hsp70) gene),多基因系统发育分析(MLSA, Multi-Locus Sequence Analysis)等;
基因组水平:基因组ANI(Average Nucleotide Identity,平均核苷酸一致性)检测、dDDH(digital DNA-DNA Hybridization,数字化DNA-DNA杂交)技术等
从保藏中心取得一株标准株,如何确定其身份?
如何保证样本与期望一致,经常是研究人员最关注的问题。
不论是保藏中心来源,还是其它机构来源,最有效的方式,便是出具有效力的菌株COA证书,包含其鉴定和溯源信息。
鉴定信息,确保菌株种属明确,溯源信息则体现了转移和传代过程,我们通常认为,5代以内的变异风险在较低水平。
细菌相比真菌,基因组紧凑,重排倾向低,无性二分裂繁殖,DNA复制度高,变异累计速度相对缓慢,只要鉴定和溯源清晰,通常和原始分离株差异不大。
关于标准菌株的定义,参照专栏《标准菌株的定义,菌株小于5代内使用的黄金法则由来?》(访问链接:https://www.huizaobio.com/article/a363.html)。
另一方面,除16S测序鉴定外,可配合其它手段进一步确认菌株身份,参上。
16S rRNA全长基因测序,能否估算样本中的菌群丰度?
在对混合样本(如粪便、土壤)进行16S扩增子测序(无论是部分区域还是全长):
每个细菌类群(如ASV或OTU)对应的测序读数(reads)数量,经过标准化(如总和为100%)后,可得到该类群在样本中的相对丰度。
相对丰度” ≠ “真实细胞比例:即使使用三代16S全长测序(如PacBio HiFi或Nanopore),偏差依然存在‘。
不同细菌基因组中16S rRNA基因拷贝数(Copy Number Variation)不同,如Bacillus:10–15 拷贝;Bifidobacterium:3–6 拷贝;Bacteroides:4–6 拷贝;Lactobacillus:1–8 拷贝(种间差异大)。
可能会出现,一个含10个拷贝的菌,即使细胞数更少,其16S信号也可能更强。
另外,DNA提取、PCR扩增效率、测序流程和数据库等差异,也会影响最终的丰度。
若研究需要接近真实的菌群丰度,建议通过宏基因组测序(Shotgun)、qPCR + 16S测序、流式细胞等技术,校正误差。
参考资料
细菌和古菌16S rRNA基因测序:原理、步骤、用途与示意图,https://www.huizaobio.com/JSZC-ZSJT/p1.html
微生物资源保藏中心的成立背景、目标使命、质量标准和其它相关事项说明, https://www.huizaobio.com/article/a365.html
16S rRNA Gene Sequencing for identification, classification and quantitation of microbes – LC Sciences – Technologies for Genomics & Proteomics Discoveries
Muhamad Rizal, N. S., et al. (2020). Advantages and Limitations of 16S rRNA Next-Generation Sequencing for Pathogen Identification in the Diagnostic Microbiology Laboratory: Perspectives from a Middle-Income Country. Diagnostics (Basel, Switzerland), 10(10), 816. https://doi.org/10.3390/diagnostics10100816
【相关资源】
名称:霍乱弧菌 | Vibrio cholerae
菌株编号:HZB368253, ATCC BAA-2163
微生物资源鉴定保藏平台
敬请关注我们,共筑健康未来!
— 武汉市灰藻生物科技有限公司团队敬上
灰藻生物:我们期待着与客户共同成长,共同创造生命科学的未来!
更新日期:2025-11-09
#创作团队
编制人:小藻 | 审稿人:小藻