细菌和古菌16S rRNA基因测序:原理、步骤、用途与示意图
来源:武汉市灰藻生物科技有限公司 浏览量:5200 发布时间:2025-11-09 13:59:29
引言
16S rRNA基因测序是一种基于扩增子的高通量测序方法,广泛用于鉴定和分类生物样本中的细菌群落。其核心在于:原核生物含有16S核糖体RNA(rRNA)基因,其序列既包含高度保守的区域,也包含具有物种特异性的可变区。前者便于设计通用引物进行扩增,后者则可用于区分不同种类。最早于1977年提出,并奠定了现代微生物系统分类的基础。
相比之下,传统培养法耗时费力,只能检测到样本中少数的微生物。而16S rRNA测序结合新一代测序技术,实现了对复杂微生物群落的快速、高效解析。正因如此,许多过去无法通过培养获得的微生物,如今已通过16S rRNA测序被成功识别并纳入数据库。
什么是16S rRNA基因?
16S rRNA基因序列是,目前研究细菌分类与系统发育,最常用的遗传标记。
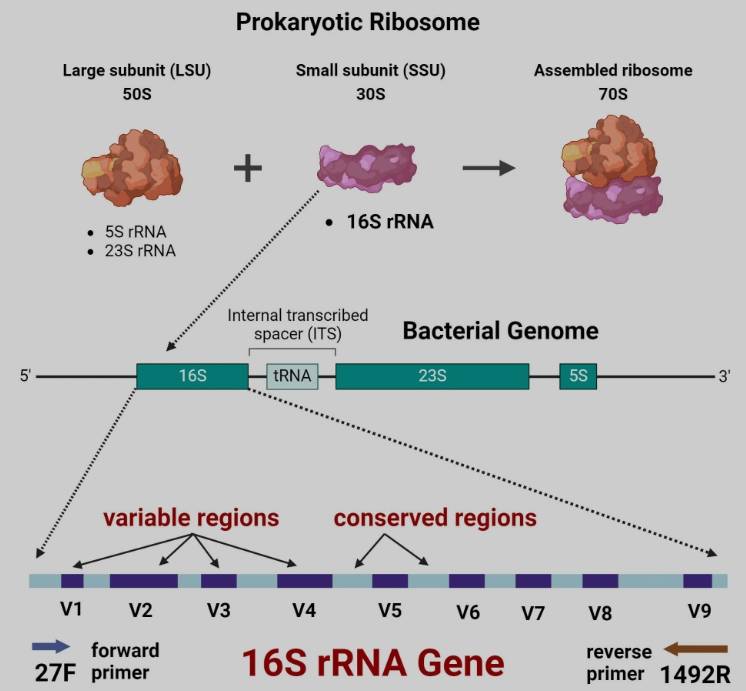
核糖体是负责蛋白质合成的分子器件,由蛋白质和核糖体RNA(rRNA)组成,包含大小两个亚基。
在原核生物中,核糖体由30S小亚基和50S大亚基构成,共同组装成完整的70S核糖体(“S”为斯维德贝格单位,表示沉降系数)。其中,30S亚基包含一条16S rRNA分子和21种蛋白质。
16S rRNA基因全长约1500个碱基对(bp),其序列结构兼具保守区与可变区:
保守区在不同细菌间高度相似,适合设计通用引物,实现对各类细菌16S基因的广泛扩增;
可变区则在不同物种间存在明显差异,可用于区分和鉴定具体的微生物种类。
整个16S rRNA基因包含9个高变区(V1–V9),这些区域的序列多样性为细菌的系统发育分析、群落组成解析以及在复杂环境中的物种识别提供了关键依据。
正因这种“保守中有变异”的特性,16S rRNA基因成为微生物生态学、临床诊断和环境监测等领域不可或缺的分子工具。
16S rRNA基因测序的原理
16S rRNA基因测序基于扩增子测序(amplicon sequencing)技术,其核心原理是:
通过靶向扩增并测序原核生物(包括细菌和古菌)基因组中,高度保守的16S rRNA基因特定区域,利用该基因序列的保守性,与变异性来实现微生物的鉴定与分类。
所有细菌和古菌均含有16S rRNA基因,其整体结构高度保守,确保了通用引物能够有效扩增来自不同物种的该基因片段;
基因内部散布的可变区(如V3–V4等)在不同物种间存在序列差异,为区分近缘微生物提供了分子依据。因此,16S rRNA基因被广泛用作,原核生物系统发育和群落分析的“分子条形码”。
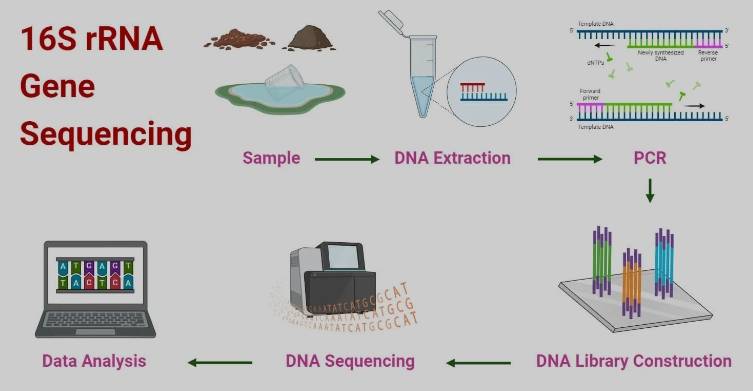
完整的测序流程包括:
16S rRNA基因测序的主要步骤
1. 样本采集
首先,从目标环境中采集微生物样本,来源包括土壤、水体、空气,或人体/动物的生物样本(如粪便、口腔拭子、皮肤等)。
不同类型的样本需采用相应的采集和保存方法,并严格避免外源污染。
在DNA提取前,常需进行预处理(如酶解、加热或物理清洗)以去除宿主细胞、腐殖酸或其他干扰物质,提高后续DNA提取的纯度与代表性。
2. DNA提取
样本采集后,需从中提取总基因组DNA。
物理破碎:如超声处理、珠磨法,用于裂解细胞壁; 化学裂解:使用去污剂(如SDS)和蛋白酶K等破坏细胞膜并降解蛋白质; 纯化:通过柱纯化、磁珠或酚-氯仿抽提等方法去除杂质,获得高纯度DNA。 提取的DNA质量直接影响后续PCR扩增效率和测序结果的可靠性。
3. PCR扩增与文库构建
旨在为测序准备标准化的DNA文库:
首先,使用针对16S rRNA基因保守区的通用引物(如341F/806R靶向V3–V4区)进行PCR扩增;引物选择和反应条件优化对避免非特异性扩增至关重要。
扩增产物随后被加上测序平台所需的接头(adapters)和样本特异性条形码(barcodes),以便多份样本混合测序后能准确区分。
文库还需经过纯化(如磁珠分选)以去除多余的引物和接头,并控制片段大小均一性,确保测序质量。
4. 测序
构建好的文库通过高通量测序平台进行测序。常用平台包括:
Illumina(如MiSeq、NovaSeq):主流选择,读长适中、通量高、错误率低; Ion Torrent、Oxford Nanopore、PacBio:适用于长读长需求,但错误率相对较高; Sanger测序:仅适用于单一菌株的16S全长测序,不适用于复杂群落。 测序过程可同时产生数万至数百万条序列读段(reads),原始数据随后进入生物信息学分析流程。
5. 数据分析
测序完成后,需借助生物信息学工具,对数据进行系统处理与解读,主要步骤包括:
质控与过滤:去除低质量读段、接头序列及非目标域(如真核生物或叶绿体)序列; 序列拼接与去噪:将双端读段合并,并生成精确的扩增子序列变体(ASV)或聚类为操作分类单元(OTU); 物种注释:将ASV/OTU与参考数据库(如 SILVA、Greengenes、EzBioCloud)比对,赋予分类学身份(界、门、纲、目、科、属,有时到种); 多样性分析:计算α多样性(样本内丰富度与均匀度)和β多样性(样本间群落差异),并进行统计与可视化。
常用分析流程包括 QIIME 2、MOTHUR 和 USEARCH/UPARSE,这些工具提供标准化流程、详细教程和用户友好界面,广泛应用于科研与临床研究。
16S rRNA测序
16S rRNA基因测序的优势
16S rRNA基因在细菌和古菌中高度保守,同时又包含足够的序列变异,足以区分不同物种。
与传统的培养方法相比,16S rRNA基因测序,无需依赖微生物的体外培养,可直接从环境或临床样本(如土壤、水体、肠道内容物等)中解析整个微生物群落,尤其能够检测大量不可培养(unculturable)的微生物——这类微生物占自然界微生物总数的90%以上,传统方法难以触及。
借助高通量测序技术(如Illumina平台),一次实验即可同时对成千上万个16S rRNA基因片段进行测序,实现对多个样本的大规模、高效率分析,显著提升了研究通量与覆盖深度。
此外,传统培养方法依赖菌落形态、生化反应等表型特征,不仅主观性强、耗时长,还容易受污染干扰。而16S rRNA基因测序采用标准化实验流程,并结合经过验证的生物信息学分析流程(如QIIME 2、MOTHUR等),内置严格的质量控制步骤,能有效减少人为误差,提高结果的可重复性与准确性。
综上,16S rRNA基因测序以其无需培养、高通量、高灵敏度和标准化程度高等优势,已成为现代微生物生态学、临床诊断和环境监测等领域的核心技术手段。
16S rRNA基因测序的局限性
分类分辨率有限:16S rRNA基因通常只能可靠地将细菌鉴定到属(genus)水平,对种(species)甚至株(strain)水平的区分能力较弱。许多亲缘关系密切的物种(如 Bacillus cereus、B. anthracis 和 B. thuringiensis;或 Escherichia coli 与部分 Shigella 菌种)在16S序列上高度相似(>99%),难以准确区分。
高序列相似性 ≠ 同一物种:即使两个菌株的16S rRNA基因序列几乎完全相同,它们在生理特性、致病性或代谢功能上仍可能存在显著差异。因此,仅凭16S数据进行物种鉴定有时会得出误导性结论。
依赖生物信息学分析:测序产生的数据量庞大且复杂,需借助专业软件(如 QIIME 2、MOTHUR、DADA2 等)进行质控、去噪、聚类和注释。这要求研究人员具备一定的生物信息学基础,否则易因参数设置不当或数据库选择偏差影响结果可靠性。
分辨率低于宏基因组测序:相比于鸟枪法宏基因组测序(shotgun metagenomics),16S测序仅靶向单一标记基因,无法提供全基因组信息,在物种分辨能力和系统发育精度上明显逊色。
无法揭示功能信息:16S rRNA测序仅反映“谁在那里”(Who is there?),但不能回答“它们能做什么”(What are they doing?)。由于不涉及功能基因,该方法无法直接推断微生物群落的代谢潜能、抗生素抗性、毒力因子等关键功能特征。
综上,16S rRNA基因测序是一种高效、经济的群落结构筛查工具,但在需要高精度物种鉴定或功能解析的研究中,常需结合全基因组测序、宏基因组学或宏转录组学等更深入的技术手段。
16S rRNA基因测序的应用
16S rRNA基因测序广泛应用于多个领域,主要用于解析复杂样本中微生物的物种组成与群落结构,从而揭示微生物多样性及其在生态系统中的功能与相互作用。以下是其主要应用场景:
1. 微生物生态学研究
通过分析土壤、水体、沉积物、空气等环境样本中的细菌和古菌群落,研究人员可评估微生物多样性、群落演替规律及环境因子对微生物分布的影响,为生态修复和生物地球化学循环研究提供依据。
2. 临床与医学微生物学
在临床领域,16S测序被用于: 探究疾病(如炎症性肠病、肥胖、糖尿病、牙周炎、败血症等)与人体微生物组失衡的关联; 鉴定传统培养方法难以检出的病原菌; 辅助诊断不明原因感染,尤其适用于无菌部位(如脑脊液、关节液)中低丰度病原体的检测; 研究医院环境中耐药菌的传播与定植动态。
3. 食品安全与发酵工业
监控发酵食品(如酸奶、泡菜、酱油、奶酪)中的有益菌群,优化生产工艺; 快速识别食源性致病菌(如 Salmonella、Listeria、Clostridium 等),保障食品卫生安全; 追踪食品加工链中的微生物污染来源。
4. 农业与植物健康
分析根际、叶际及土壤微生物组,挖掘促进植物生长、抗病或固氮的功能菌群; 指导微生物肥料或生物农药的开发; 评估耕作方式、化肥使用或转基因作物对土壤微生态的影响。
5. 人类微生物组研究
作为“人类微生物组计划”(Human Microbiome Project, HMP)的核心技术之一,16S测序系统描绘了人体各部位(肠道、口腔、皮肤、阴道等)的正常菌群图谱,推动了对微生物与免疫、代谢、神经发育等健康过程关系的理解。
6. 新物种发现与系统分类
通过比对未知菌株的16S序列与数据库,可初步判断其是否为潜在新种。当序列相似度低于98.7%(通常作为种界定阈值)时,可能提示新分类单元的存在,为进一步分类学研究提供线索。
典型应用
人类微生物组计划(HMP):全面刻画人体共生微生物组成;
全球海洋采样远征(Global Ocean Sampling Expedition):揭示海洋微生物的全球分布与多样性;
土壤微生物组研究:评估土地利用变化或气候变化对土壤生态的影响;
医院耐药菌监测:追踪多重耐药菌在病房或ICU中的传播路径;
污水处理系统监控:优化工艺参数,提升有机物降解与脱氮除磷效率。
总之,16S rRNA基因测序凭借其高通量、低成本和无需培养的优势,已成为连接基础研究与实际应用的重要桥梁,在环境、健康、农业和工业等多个领域持续发挥关键作用。
参考资料
16S and ITS rRNA Sequencing | Identify bacteria & fungi with NGS (illumina.com)
16S rRNA Gene Sequencing for identification, classification and quantitation of microbes – LC Sciences – Technologies for Genomics & Proteomics Discoveries
Muhamad Rizal, N. S., et al. (2020). Advantages and Limitations of 16S rRNA Next-Generation Sequencing for Pathogen Identification in the Diagnostic Microbiology Laboratory: Perspectives from a Middle-Income Country. Diagnostics (Basel, Switzerland), 10(10), 816. https://doi.org/10.3390/diagnostics10100816
【相关资源】
名称:霍乱弧菌 | Vibrio cholerae
菌株编号:HZB368253, ATCC BAA-2163
微生物资源鉴定保藏平台
敬请关注我们,共筑健康未来!
— 武汉市灰藻生物科技有限公司团队敬上
灰藻生物:我们期待着与客户共同成长,共同创造生命科学的未来!
更新日期:2025-11-09
#创作团队
编制人:小藻 | 审稿人:小藻