分子实验笔记14:不依赖连接酶的克隆(Ligation Independent Cloning, LIC)
来源:武汉市灰藻生物科技有限公司 浏览量:230 发布时间:2026-03-24 20:57:31
概述(Overview)
不依赖连接酶的克隆(Ligation Independent Cloning, LIC)省去了传统克隆方法中耗时的连接(ligation)步骤。
在传统克隆中,短片段间的碱基配对(通常仅4 bp)不足以在转化和复制过程中维持质粒结构的稳定性。
而LIC利用长单链突出端(long overhangs,通常10–12 bp)形成稳定的片段间结合,从而无需连接即可直接进行转化。
最终,这种虽已退火但存在切口(nicked)的环状载体可在宿主细胞的DNA复制过程中被修复。
LIC常用T4 DNA聚合酶(T4 DNA polymerase),该酶兼具外切酶(exonuclease)与聚合酶(polymerase)活性,可根据特定序列生成长度可控的单链突出端。
用于LIC的空载体通常采用II型限制性内切酶(type II restriction enzymes,如BsaI),这类酶在其识别位点外一定距离处切割DNA。
使用单一酶即可产生多种不同的突出端,并且在最终产物中**完全去除酶切位点**,实现“无痕克隆”(no "cloning scars")。
载体中常包含一段“填充序列”(stuffer sequence),便于通过电泳将线性化载体与酶切副产物分离;
该序列还可作为反向筛选标记(counter-selection marker),例如图中所示的sacB基因,可有效抑制非重组克隆的生长。
LIC专用载体的生产商通常会提供一段同源序列(homologous sequence),需将其整合到PCR引物的5'端。下文以质粒pNIC28-Bsa4为例,说明LIC实验设计流程。
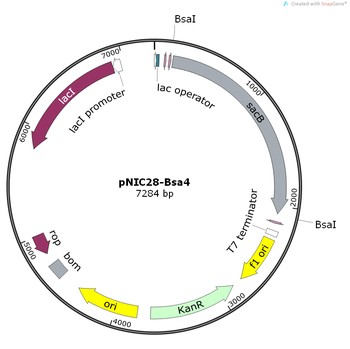
图1:pNIC28-Bsa4
操作流程(Protocol)
步骤1:引物设计(Design Your Primers)
LIC引物设计通常只需将载体厂商提供的前导序列(leader sequence)与目标基因序列融合即可,注意保持与起始密码子或标签序列(如His-tag)的阅读框一致(in frame)。
引物长度取决于T4 DNA聚合酶的“回切”(chew back)反应机制:反应体系中仅加入一种脱氧核苷三磷酸(dNTP),可终止外切酶活性并触发聚合酶活性。
本例中使用dGTP,因此T4 Pol会从3'端逐个切除碱基,直至遇到第一个G(图中蓝色标出),此时酶会重新加上该G并停滞。因此,引物必须以紧随其后的T开头,以确保最终组装无缺口。
通常,引物包含15 bp同源序列 + 至少18 bp模板特异性序列。正向(5')与反向(3')引物的前导序列不同,但均遵循相同原则——即与酶切位点下游第一条链上首个G之前的序列互补。
为简化说明,此处仅展示5'引物设计。

图2:引物设计
注意:建议使用在线引物设计工具,确保PCR引物的熔解温度(Tm)在50–60°C之间。
步骤2:线性化载体(Linearize Vector)
本例中,载体经BsaI酶在两个位点切割,移除“填充序列”及sacB反向筛选标记(参见我们的限制性酶切实验方案)。
酶切完成后,需通过琼脂糖凝胶电泳分离线性化载体,并进行凝胶纯化。酶切后载体末端结构如下所示:

图3:线性化载体
注意:为确保后续反应的离子强度适宜,请使用无菌水(sterile water)而非TE缓冲液洗脱纯化的酶切产物和PCR产物。
步骤3:制备载体单链突出端(Create Vector Overhangs)
按照厂商说明书,用T4 DNA聚合酶处理线性化载体,使其3'端发生“回切”。
本反应体系中仅加入dGTP(不添加其他dNTPs),促使酶持续发挥外切活性,直至遇到序列中的第一个G。
由于聚合酶活性优先于外切活性,酶会重新加入该G并停滞,从而形成确定长度的单链突出。
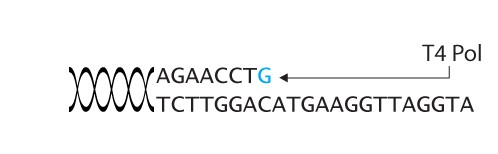
图4:制备载体单链突出端
典型T4 Pol反应体系如下(总体积40 μl):
| 体积 (μl) | 试剂 (Reagent) | 终浓度 (Final Conc.) |
|---|---|---|
| 4 | 10X Buffer | 1X |
| 20–30 | 洗脱DNA (Eluted DNA) | 10–50 ng/μl |
| 1 | dGTP (100 mM) | 2.5 mM |
| 2 | DTT (100 mM) | 5 mM |
| 1 | BSA (10 μg/μl) | 0.25 μg/μl |
| 1 | T4 DNA polymerase | 0.075 units/μl |
| 补足至40 μl | 无菌dH₂O (sterile dH₂O) | — |
混匀各组分(最后加入T4酶),室温孵育30分钟,随后75°C加热20分钟以灭活酶活性。
步骤4:PCR扩增插入片段(Amplify Insert by PCR)
按所用高保真聚合酶说明书进行插入片段的PCR扩增。**关键步骤**:必须彻底去除PCR产物中残留的游离dNTPs,否则会干扰下一步T4 Pol的外切活性。推荐通过凝胶纯化实现此目的。

图5:PCR扩增插入片段
步骤5:制备插入片段单链突出端(Create Insert Overhangs)
用T4 DNA聚合酶处理纯化的PCR产物,但本次反应中加入的是dCTP(而非dGTP),以匹配载体酶切后5'端暴露的第一个碱基(图中蓝色所示)。
反应同样可通过75°C加热20分钟终止。

图6:制备插入片段单链突出端
步骤6:退火与转化(Anneal and Transform)
将处理后的载体与插入片段按1:2 或 1:3 的摩尔比混合,每管退火反应使用20–50 ng载体,总体积应小于5 μl(不额外加水)。
建议固定载体量,设置多个不同插入片段浓度的反应,以提高成功率。同时设立“仅载体”对照(用水替代插入片段)。
室温孵育5分钟,加入1 μl 25 mM EDTA(螯合Mg²⁺以彻底终止酶活),再室温孵育5分钟。退火产物即可用于转化。按标准流程,取1–2 μl退火反应液转化感受态细胞。

图7:退火与转化
参考文献
https://www.addgene.org/
敬请关注灰藻生物,共筑健康未来!
— 武汉市灰藻生物科技有限公司团队敬上
灰藻生物:我们期待着与客户共同成长,共创生命科学的美好未来!
更新日期:2026-03-27
编制人:磊子
审稿人:小藻