分子实验笔记12:基于PCR的质粒克隆(Plasmid Cloning by PCR)
来源:武汉市灰藻生物科技有限公司 浏览量:298 发布时间:2026-03-23 19:38:32
背景(Background)
基于PCR的克隆(PCR-based cloning)具有极高的灵活性,几乎可将任意DNA片段插入所选的载体骨架(backbone vector)中,限制极少。
其基本原理是:在扩增目标DNA片段的同时,在其两端引入限制性酶切位点(restriction sites),以便后续将其高效克隆至目标质粒中。
本示例将演示,如何将一个cDNA从原始载体,转移至更适合功能研究的新载体。
如下图所示,我们将在目标基因(Your Gene of Interest, YGOI)两端分别添加EcoRI和NotI酶切位点,以便将其连接至受体质粒(recipient plasmid)。
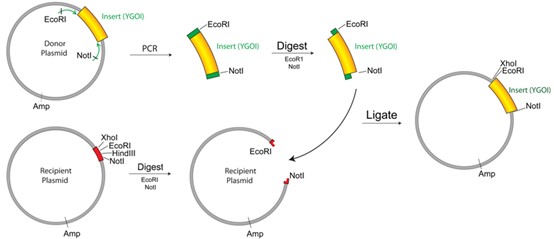
图1:载体构建示例
PCR克隆引物设计(Designing Primers for PCR Based Cloning)
用于分子克隆的PCR引物通常包含以下三部分:
- 前导序列(Leader Sequence):位于引物5'端的额外碱基(通常3–6 bp),有助于提高限制酶切割效率;
- 限制性酶切位点(Restriction Site):所选的克隆用酶切位点(通常6–8 bp);
- 杂交序列(Hybridization Sequence):与目标序列互补结合的区域(通常18–21 bp)。
选择限制酶时,应使用DNA分析工具(如Addgene Sequence Analyzer)确认目标序列中是否存在该酶切位点。理想情况下,所选酶应满足:
- 在插入片段(insert)内部无切割位点;
- 在受体质粒的多克隆位点(Multiple Cloning Site, MCS)中有且仅有该位点;
- 加分项:两种能在同一种缓冲液(same buffer)中高效工作的酶更便于双酶切操作。
本例中,我们将使用EcoRI和NotI将cDNA插入受体质粒。为确保插入方向正确,需根据MCS结构,将上游酶切位点(EcoRI)置于正向引物(forward primer)5'端,下游酶切位点(NotI)置于反向引物(reverse primer)5'端。
接下来,需分析待扩增的DNA序列并设计特异性引物。下图展示了开放阅读框(ORF)两端序列及其在引物设计中的应用:
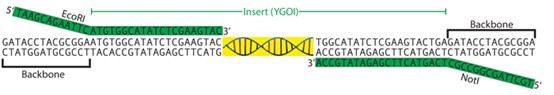
图2:引物设计
由于克隆的是完整ORF,我们需从起始密码子(ATG)扩增至终止密码子(本例为TGA)。若以质粒DNA为模板(而非基因组DNA或cDNA文库),18–21 bp的杂交序列通常足以保证特异性和PCR效率。
因此,正向引物的杂交序列为:5'-ATGTGGCATATCTCGAAGTAC-3'
在其5'端添加EcoRI位点(GAATTC),得到:5'-GAATTCATGTGGCATATCTCGAAGTAC-3'
需注意:许多限制酶在DNA线性末端切割效率较低(详见NEB说明)。因此,建议在酶切位点上游额外添加3–6个碱基以提升切割效率。这些碱基可任意选择,但应避免形成引物内部发夹结构(hairpin)。本例中添加TAAGCA,最终正向引物序列为:5'-TAAGCAGAATTCATGTGGCATATCTCGAAGTAC-3'
反向引物设计类似,但需使用目标序列的反向互补链(reverse complement)以实现PCR扩增。我们取ORF末端18 bp(含终止密码子):5'-TGGCATATCTCGAAGTACTGA-3'
依次添加NotI位点(GCGGCCGC)和前导序列TAAGCA,得:5'-TGGCATATCTCGAAGTACTGAGCGGCCGCTAAGCA-3'
再对其取反向互补(可使用在线工具,如SMS Reverse Complement),最终反向引物序列为:5'-TGCTTAGCGGCCGCTCAGTACTTCGAGATATGCCA-3'
实验流程(Experimental Procedure)
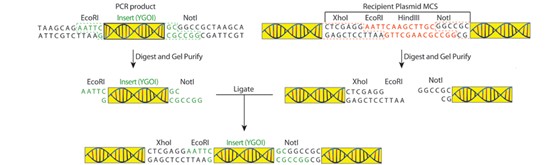
图3:试验流程
1. 进行PCR并纯化产物
- 使用高保真Taq DNA聚合酶(high-fidelity Taq polymerase)扩增插入片段,以最大限度减少突变。产物越长,对聚合酶保真度要求越高。
- 退火温度应基于引物中杂交序列部分(即结合ORF的18–21 bp)的熔解温度(Tm)设定,而非整个引物的Tm。
- 若模板为质粒或简单DNA,非特异性扩增风险低,退火温度范围可较宽;若从基因组DNA、cDNA文库或通过RT-PCR扩增,则需延长引物、提高Tm以增强特异性。
- 使用试剂盒(如QIAquick PCR Purification Kit)纯化PCR产物,所得DNA即可用于后续酶切。
2. 酶切DNA
- 分别对PCR产物和受体质粒进行限制性酶切。由于后续凝胶纯化会损失DNA,建议使用全部PCR产物及1 μg受体质粒。
- 为确保受体质粒被双酶完全切割,酶切反应应至少进行4小时,过夜更佳。
- 重要提示:若仅使用单酶切,或所用酶产生兼容黏性末端/平末端,则必须使用磷酸酶(phosphatase)处理线性化载体,防止其自连。常用酶包括小牛碱性磷酸酶(CIP)或虾碱性磷酸酶(SAP),处理可在连接前或凝胶纯化前进行,具体依所选磷酸酶说明书而定。
3. 凝胶纯化插入片段与载体
- 将酶切产物进行琼脂糖凝胶电泳,并切胶回收目标条带。为获得清晰、分离良好的条带,建议:
- 除DNA Marker外,建议同时点样未酶切的载体作为对照,便于排查酶切异常。
- PCR克隆中,电泳验证尤为关键——可确认PCR产物大小是否正确、条带强度是否足够(反映扩增效率与DNA得量)。
- 切胶纯化后,务必测定回收DNA的浓度。
4. 将插入片段连接至载体
- 标准连接反应总DNA量建议约100 ng。理想摩尔比为载体:插入片段 = 1:3。因两者长度不同,仅凭浓度难以精确计算,建议对每个构建设置两组不同比例的连接反应以优化条件。
- 必须设置阴性对照:例如,仅连接受体质粒(不加插入片段),用于评估载体自连或未切尽产生的背景克隆数量。
5. 转化(Transformation)
- 按感受态细胞说明书进行转化。常规克隆中,可将1–2 μL连接产物转化至DH5α或TOP10等化学感受态细胞。
- 若总DNA量极低(<1 ng)或难以获得菌落,建议使用高转化效率感受态细胞;若最终质粒较大(>10 kb),推荐使用电转化感受态细胞(electro-competent cells)。
- 转化后菌落数是初步判断实验成败的关键:“载体+插入片段”平板的菌落数应显著高于“仅载体”对照平板。后者反映背景水平——即非正确重组子的预期数量。
- 若“仅载体”对照菌落过多,可增设两组对照:一组加连接酶,一组不加。若不加连接酶仍有菌落,说明存在未切尽的环状空载体;若仅加连接酶组有大量菌落,则为载体自连所致。
- 若无任何菌落,应进行阳性对照(如转化已知质粒)验证感受态细胞活性,并尝试调整载体与插入片段的比例。
6. 获取重组质粒
- 挑取单菌落(根据对照平板背景高低,通常挑3–10个),接种过夜培养用于质粒提取。
- 取100–300 ng纯化质粒,用克隆所用的限制酶进行诊断性酶切(diagnostic digest),电泳检测。正确克隆应显示两条带:一条为载体大小,另一条为插入片段大小。
7. 测序验证(Verify your Plasmid by Sequencing)
基于PCR的克隆比传统酶切克隆具有更高的突变风险。PCR扩增的错误率因聚合酶而异,范围约为每500 bp至每1000万 bp出现1个错误。因此,无论使用何种高保真聚合酶,最终构建的质粒都必须进行测序验证。
参考文献
https://www.addgene.org/
敬请关注灰藻生物,共筑健康未来!
— 武汉市灰藻生物科技有限公司团队敬上
灰藻生物:我们期待着与客户共同成长,共创生命科学的美好未来!
更新日期:2026-03-23
编制人:磊子
审稿人:小藻