如何进行微生物序列分析?FASTA和BLAST理论介绍,及公共数据库16S序列分析操作教程
来源:武汉市灰藻生物科技有限公司 浏览量:1030 发布时间:2025-10-30 21:41:07
引言
在基因组学时代,随着DNA和蛋白质序列数据呈指数级增长,如何解读这些生命“密码”成为了关键。生物信息学应运而生,其核心任务之一就是序列分析。
本文将深入探讨序列分析的基石——序列比对,并回顾两大关键工具FASTA与BLAST的发展与革新,从而构建起从理论到实践的完整知识框架。
第一章:序列比对——一切分析的基石
序列比对是生物信息学中最基本、最核心的操作,它通过排列两个或多个序列(核苷酸或氨基酸),来识别它们之间的相似性区域。
1.1 为什么需要序列比对?
序列的相似性往往暗示着功能、结构或进化上的关联性。
通过比对,我们可以:
• 鉴定未知序列:通过与已知序列数据库对比,推测新序列的功能。
• 推断进化关系:构建系统发育树,理解物种或基因间的亲缘关系。
• 识别保守域与模体:找到对功能至关重要的关键区域。
• 预测蛋白质结构:序列相似性高的蛋白质,其三维结构也往往相似。
1.2 比对的核心类型
• 全局比对:试图将两个序列的全长进行对齐,适用于长度和整体相似度都非常接近的序列。Needleman-Wunsch算法是其主要实现方法。
• 局部比对:只寻找序列间相似度最高的局部区域进行对齐,适用于在长序列中寻找保守域或模体。Smith-Waterman算法是其主要实现方法。
# 核心区别:全局比对要求“整体最优”,而局部比对寻找“局部最优”。在实际研究中,由于基因结构域的模块化,局部比对的应用更为广泛。
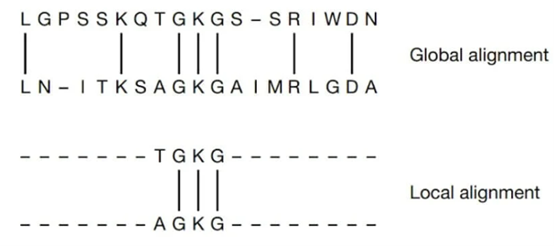
图1、 两个序列的全局与局部比对
1.3 两两序列比对的方法
1. 点阵法:
点阵法(又称点图法)是一种图形化的序列比对方法,通过在二维矩阵中比较两个序列来实现。
具体做法是:将两个待比较的序列分别置于矩阵的横轴和纵轴上,然后逐一扫描一个序列中的每个残基,与另一个序列的所有残基进行比较。
若两个残基匹配,则在对应位置标记一个点;否则留空。
• 如果两个序列高度相似,点图将沿主对角线呈现一条清晰的直线;
• 若相似性较低,则点分布更分散,对角线不明显;
• 点图还可用于发现单个序列内部的重复元件:主对角线上下出现的短平行线即表示重复结构。
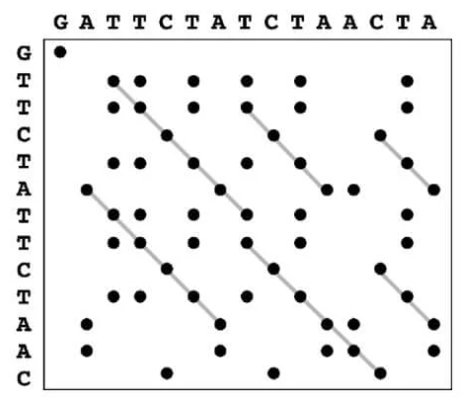
图2、 使用点图法比较两个序列的示例
2. 动态规划法:
动态规划法通过比较两个蛋白质或核酸序列中所有可能的字符组合,找出最优比对结果。
该方法既可用于全局比对,也可用于局部比对:
• 全局比对采用 Needleman-Wunsch 算法;
• 局部比对采用 Smith-Waterman 算法。
动态规划法包含以下三个步骤:
• 评分矩阵初始化:构建一个二维矩阵,将两条待比对序列分别置于顶部和左侧。矩阵左上角初始化为0,并根据空位罚分规则进行初始化。
• 填充矩阵:根据评分矩阵逐行填充每个单元格。
• 对于核苷酸序列,匹配得正分,错配得负分;
• 对于氨基酸序列,则使用 BLOSUM 或 PAM 等专业评分矩阵。
算法从左上角开始,逐行向右下角推进,每个单元格填入当前可能的最大得分。
• 回溯(Traceback):填充完成后,从右下角开始回溯至左上角,寻找得分最高的路径,从而确定最优比对结果。
3. 启发式方法:
为了在速度和灵敏度之间取得平衡而开发的“捷径”算法。它们不保证找到绝对最优解,但能在极短时间内找到非常优异的近似解。这正是FASTA和BLAST成功的关键。
1.4 多序列比对
当需要比较三个及以上序列时,就进入了多序列比对的领域。它对于识别保守区域、构建系统发育树至关重要。
由于计算复杂度极高,通常采用启发式方法,如:
• A. 穷举算法(Exhaustive algorithms)
• 穷举法尝试同时评估所有可能的比对方案。
• 类似于两两比对中的二维动态规划矩阵,多重比对需要构建一个 N 维搜索矩阵(N 为序列数量)。
• 随着序列数量增加,计算时间和内存需求呈指数级增长,因此该方法仅适用于少于10条短序列的小规模数据集。
• 对于大规模数据,通常采用启发式方法以提高效率。
B. 启发式算法(Heuristic algorithm)
i. 渐进法(Progressive method)
又称基于树的算法,通过逐步组装的方式构建多重比对。
步骤如下:
• 使用 Needleman–Wunsch 全局比对法对所有序列进行两两比对,获得相似性得分;
• 将得分转换为进化距离,构建距离矩阵;
• 利用邻接连接法(neighbor-joining)从距离矩阵生成引导树(guide tree);
• 按照引导树的拓扑结构,从最相近的两个序列开始,逐步加入其他序列,最终完成多重比对。
常用工具包括 Clustal 和 T-Coffee。
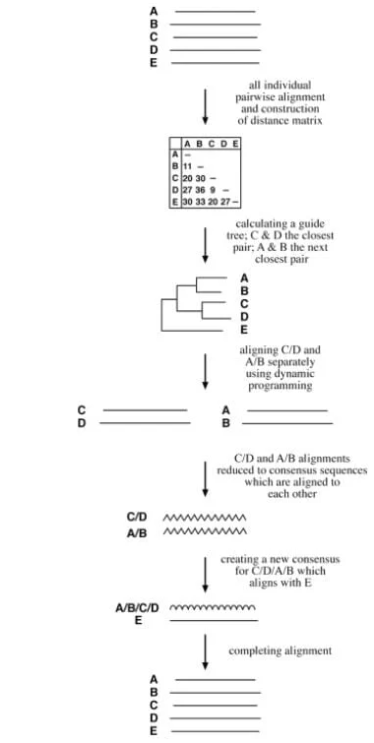
图3、渐进比对流程
ii. 迭代法(Iterative Method)
迭代法通过反复优化初始的次优比对,逐步逼近最优解。
首先进行初步两两比对以构建系统发育树,并据此赋予权重;
随后识别含空位的区域并反复调整,以提高比对得分;
每次迭代后,基于新比对结果重新计算树、权重和比对,直至得分不再提升。
PRRN 是一个基于迭代法的在线比对程序。
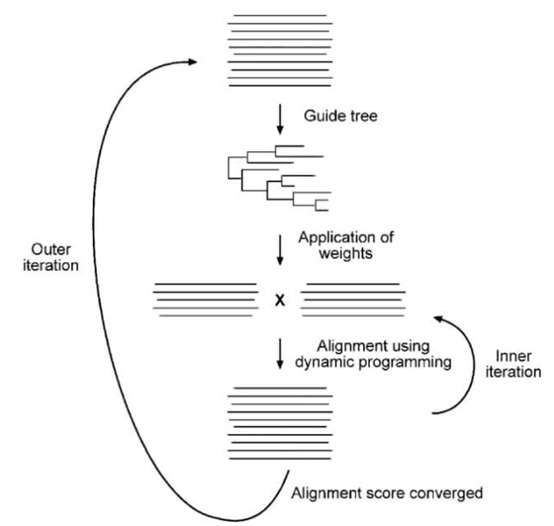
图4、PRRN 的迭代比对流程
iii. 基于模块(Block-based)的方法
渐进法和迭代法主要基于全局比对,在处理长度差异大、高度分歧的序列时,难以有效识别保守结构域或基序(motif)。
此时需采用基于局部比对的策略。
模块法正是此类方法之一:它识别所有序列共有的无空位比对片段(称为“模块”或“blocks”),特别适用于功能域分析。
第二章:FASTA——开创性的实践
在动态规划法因计算资源限制而难以实用化之时,FASTA的出现标志着序列数据库相似性搜索的开端。
2.1 FASTA是什么?
FASTA(或称 FastA,全称为 “Fast-All”)是最早被广泛使用的数据库相似性搜索工具之一。
它是一种序列比对工具,可将输入的核苷酸或蛋白质序列与数据库中的序列进行比对。FASTA 最初由 David J. Lipman 和 William R. Pearson 于 1985 年开发,此后不断改进,已广泛应用于多种场景。
由 FASTA 程序衍生出的用于表示核苷酸或蛋白质序列的纯文本文件格式(即 FASTA 格式),如今已成为生物信息学领域的标准格式。许多其他序列数据库搜索工具也采用 FASTA 文件格式。

图5、FASTA 格式示例。图片来源:NCBI
2.2 FASTA如何工作?——四步法
FASTA采用了一种多步骤的启发式策略,在保证精度的同时大幅提升速度:
步骤 1:识别高相似区域(Identifying Regions)
首先,FASTA 为查询序列构建一个查找表(lookup table),也称为哈希表(hashing step)。
具体做法是将查询序列分割成若干短片段,称为 k-tuples(k 元组):
• 对于蛋白质序列,k 通常为 2;
• 对于 DNA 序列,k 通常为 6。
增大 k 值可减少背景噪声(即随机匹配的 k-tuple 数量),从而提高搜索效率和特异性。
构建好查找表后,FASTA 扫描数据库,寻找与查询序列中 k-tuple 匹配的位置。这些匹配在二维矩阵中表现为对角线。程序会保留匹配密度最高的前10条对角线,作为潜在的高相似区域。
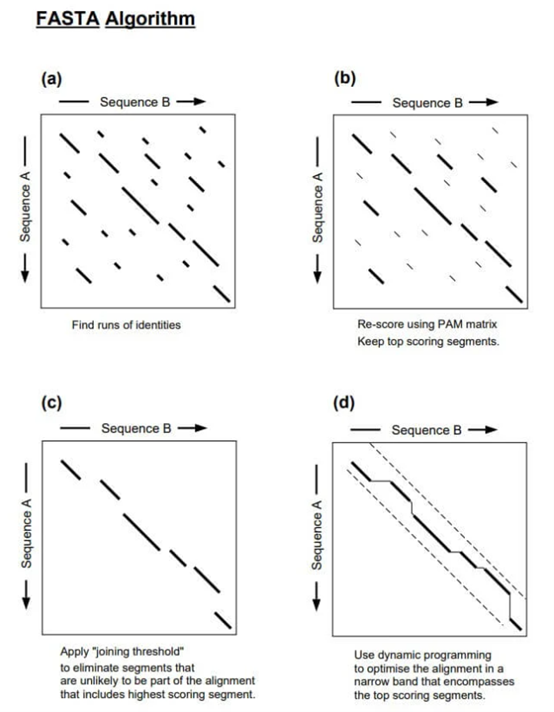
图6、FASTA 算法示意图(Barton, G.J., 1996)
步骤 2:重新打分(Re-Scoring)
在第二步中,FASTA 对这10条最佳对角线使用合适的打分矩阵进行重新评估:
• 蛋白质比对常用 BLOSUM50 或 PAM 矩阵;
• DNA 比对则使用单位矩阵(identity matrix,即匹配得1分,错配得0分)。
对每条对角线,找出其中得分最高的子区域,这些子区域称为初始区域(initial regions)。
步骤 3:连接阈值(Joining Threshold)
接下来,FASTA 设置一个打分阈值(joining threshold),用于排除不太可能属于最终比对的片段。
数据库中的序列根据初始得分进行排序,只有得分高于阈值的区域才会被保留,并尝试将相邻的高分区域连接起来。
在此过程中,FASTA 允许在对角线之间引入空位(gaps),并施加空位罚分。最终的带空位比对得分 = 区域得分总和 − 空位罚分。该得分用于对数据库序列按相似性排序。
步骤 4:最终比对(Final Alignment)
最后,FASTA 使用带状 Smith-Waterman 算法(banded Smith-Waterman algorithm)对初步比对结果进行精细化优化。
这是一种动态规划算法,能计算出最优比对得分(optimal score, 简称 opt),并用于后续的统计显著性评估。
2.3 FASTA的贡献与特点
• 开创性:首次让科研人员能够快速搜索大型数据库。
• 统计评估:除了E值(期望值,表示随机匹配的概率),FASTA还提供了Z值,用于衡量匹配得分相对于整个数据库得分分布的显著性。
• 程序家族:FASTA发展出一个工具家族,如FASTX(允许翻译DNA并比对蛋白)、TFASTX等,应对不同类型的搜索需求。
第三章:BLAST——效率的革命
尽管FASTA已经很快,但数据的爆炸式增长呼唤更快的工具。1990年,BLAST的横空出世,将数据库搜索的速度和可用性提升到了一个全新的高度,成为了至今最主流的生物信息学工具。
3.1 BLAST是什么?
BLAST(Basic Local Alignment Search Tool)由Stephen Altschul等人开发。其设计哲学与FASTA类似,但在算法细节上进行了优化,实现了速度的飞跃。
图7、BLAST,图片来源:NCBI
3.2 BLAST如何工作?——种子延伸策略
BLAST的工作流程更为简洁高效:
BLAST 通过将查询序列与数据库中的序列进行比对,寻找相似区域。它采用启发式算法(heuristic approach),在保证较高灵敏度的同时显著提升搜索速度。
BLAST 的比对过程主要包括以下四个步骤:
步骤 1:构建词表(Seeding)
BLAST 首先将查询序列分割成若干短片段,称为“词”(words),并构建一个查找表(lookup table),这一步也称为“种子生成”(seeding)。
• 对于蛋白质序列,每个词通常由 3 个氨基酸组成;
• 对于DNA 序列,每个词通常为 11 个核苷酸长。

图8、BLAST 工作原理——步骤 1。图片来源:NLM, NCBI
步骤 2:数据库扫描(Hit Finding)
BLAST 在数据库中搜索包含与查询词完全相同的序列片段。这一步用于快速筛选出可能与查询序列相关的候选数据库序列。

图9、BLAST 工作原理——步骤 2,图片来源:NLM, NCBI
步骤 3:词匹配打分(Scoring)
对匹配的词使用替换矩阵(substitution matrix)进行打分:
• 蛋白质比对常用 PAM(Percent Accepted Mutations)或 BLOSUM(Blocks Substitution Matrix)矩阵;
• DNA 比对则采用简单的匹配/错配打分(如匹配 +1,错配 -1)。
只有得分超过预设阈值的词才被视为有效“种子”,用于后续扩展。
步骤 4:延伸与高分片段对(HSP)生成
BLAST 从每个有效种子开始,向两端延伸比对,同时持续计算比对得分(使用相同的替换矩阵)。
如果延伸过程中得分因错配或空位而下降至阈值以下,则停止延伸。
最终得到的无空位局部比对区域称为 高分片段对(High-scoring Segment Pair, HSP)。
此外,BLAST 还会为每个 HSP 计算一个统计显著性值,即 E 值(Expect value):
• E 值表示在随机情况下获得当前或更高得分的期望次数;
• E 值越小,比对结果越显著(例如 E 小于 1e-5 通常被认为具有生物学意义)。
3.3 BLAST为何如此成功?
• 无与伦比的速度:其“种子延伸”算法极其高效。
• 用户友好:提供了清晰的网页界面和易于解读的格式化结果。
• 高度的敏感性与灵活性:通过调整词长、打分矩阵等参数,可以平衡速度与灵敏度。
• 强大的工具生态:根据不同的查询序列和数据库类型,BLAST衍生出五大核心变体:
o BLASTN:核酸 vs 核酸库
o BLASTP:蛋白 vs 蛋白库
o BLASTX:核酸(翻译后) vs 蛋白库
o TBLASTN:蛋白 vs 核酸(翻译后)库
o TBLASTX:核酸(翻译后) vs 核酸(翻译后)库
总结
回顾这三部分内容,我们可以清晰地看到一个承前启后、不断优化的技术发展脉络:
1. 理论基础
序列比对提供了整个领域的数学和概念基础。它告诉我们什么是“相似”,以及如何通过动态规划等方法找到“最优”的相似性。这是“道”。
2. 开创性实践
FASTA是将理论应用于大规模实际问题的一次伟大实践。它通过启发式方法(k-tuple、重新评分、连接),在可接受的时间内实现了数据库搜索,是“术”的突破。
3. 效率革命
BLAST在FASTA的基础上,对“术”进行了极致优化。其“种子延伸”策略更加激进和高效,结合友好的界面和强大的统计(E值),最终使其成为全球生物学实验室的标配工具,完成了从“可用”到“好用”的革命。
如何在FASTA和BLAST之间选择?
• 默认选择BLAST:在绝大多数情况下,BLAST因其速度和便捷性是首选。它的E值统计非常直观。
• 当BLAST无果时尝试FASTA:由于算法差异,FASTA有时能发现BLAST遗漏的微弱相似性(特别是在涉及复杂模式或远缘同源时)。
• 特定场景:FASTA的某些特定程序(如FASTX/Y在解决含有移码错误的序列时)可能更有优势。
操作教程
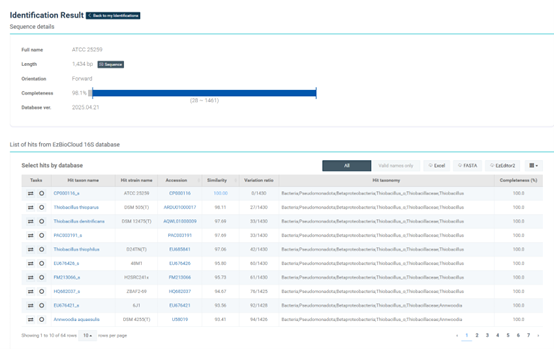
如何在NCBI或EZBio-cloud比对菌种16S序列,以ATCC 25259 脱氮硫杆菌 Thiobacillus denitrificans为例
一、NCBI:
• 打开NCBI首页(https://www.ncbi.nlm.nih.gov/)
• 点击右侧“BLAST”
• 点击最左侧“Nucleotide BLAST”
• 在“Enter accession number(s), gi(s), or FASTA sequence(s) ”中输入测序公司提供的序列,或下方直接选择相应的【seq】文件,随后点击最下方“BLAST”按钮
• 等待1分钟左右
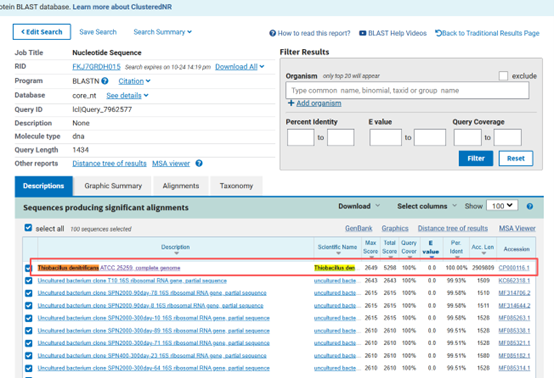
• 分析比对结果。
二、EZBio-Cloud:
• 打开EZBio-Cloud:首页(https://www.ezbiocloud.net/)
• 点击“16S-based ID”
• 点击“Identify new sequence”
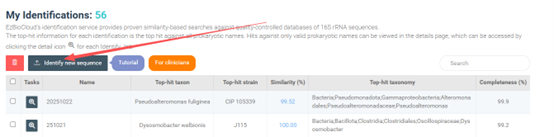
• 在弹出的框内输入序列名字、序列,点击“NEXT”
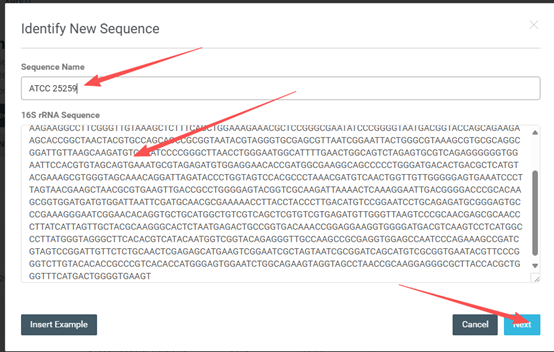
• 点击“Submit”
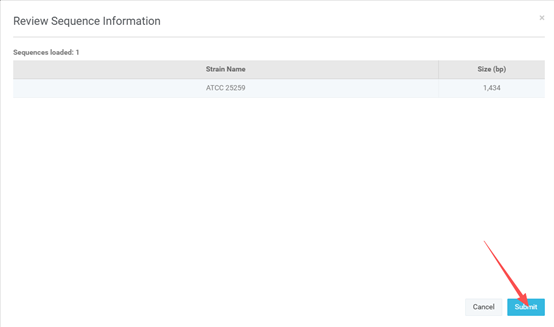
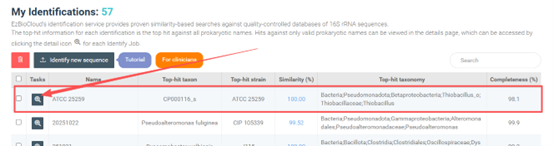
• 等待1分钟左右,进行结果分析。
参考文献
1、https://microbenotes.com/local-global-multiple-sequence-alignment/
2、https://microbenotes.com/fasta/
3、https://microbenotes.com/blast-bioinformatics/
相关产品
HZB357313=ATCC 25259:脱氮硫杆菌 | Thiobacillus denitrificans
16S序列:
ATGCAAGTCGAACGGCAGCACGGGAGCTTGCTCCTGGTGGCGAGTGGCGAACGGGTGAGTAATGCGTCGGAACGTACCG
AGTAATGGGGGATAACGCACCGAAAGGTGTGCTAATACCGCATACGCCCTGAGGGGGAAAGTGGGGGACCGCAAGGCCT
CACGTTATTCGAGCGGCCGACGTCTGATTAGCTAGTTGGTGGGGTAAAGGCCTACCAAGGCGACGATCAGTAGCGGGTC
TGAGAGGATGATCCGCCACACTGGGACTGAGACACGGCCCAGACTCCTACGGGAGGCAGCAGTGGGGAATTTTGGACAA
TGGGCGAAAGCCTGATCCAGCCATTCCGCGTGAGTGAAGAAGGCCTTCGGGTTGTAAAGCTCTTTCAGCTGGAAAGAAA
CGCTCCGGGCGAATATCCCGGGGTAATGACGGTACCAGCAGAAGAAGCACCGGCTAACTACGTGCCAGCAGCCGCGGTA
ATACGTAGGGTGCGAGCGTTAATCGGAATTACTGGGCGTAAAGCGTGCGCAGGCGGATTGTTAAGCAAGATGTGAAATC
CCCGGGCTTAACCTGGGAATGGCATTTTGAACTGGCAGTCTAGAGTGCGTCAGAGGGGGGTGGAATTCCACGTGTAGCA
GTGAAATGCGTAGAGATGTGGAGGAACACCGATGGCGAAGGCAGCCCCCTGGGATGACACTGACGCTCATGTACGAAAG
CGTGGGTAGCAAACAGGATTAGATACCCTGGTAGTCCACGCCCTAAACGATGTCAACTGGTTGTTGGGGGAGTGAAATC
CCTTAGTAACGAAGCTAACGCGTGAAGTTGACCGCCTGGGGAGTACGGTCGCAAGATTAAAACTCAAAGGAATTGACGG
GGACCCGCACAAGCGGTGGATGATGTGGATTAATTCGATGCAACGCGAAAAACCTTACCTACCCTTGACATGTCCGGAA
TCCTGCAGAGATGCGGGAGTGCCCGAAAGGGAATCGGAACACAGGTGCTGCATGGCTGTCGTCAGCTCGTGTCGTGAGA
TGTTGGGTTAAGTCCCGCAACGAGCGCAACCCTTATCATTAGTTGCTACGCAAGGGCACTCTAATGAGACTGCCGGTGA
CAAACCGGAGGAAGGTGGGGATGACGTCAAGTCCTCATGGCCCTTATGGGTAGGGCTTCACACGTCATACAATGGTCGG
TACAGAGGGTTGCCAAGCCGCGAGGTGGAGCCAATCCCAGAAAGCCGATCGTAGTCCGGATTGTTCTCTGCAACTCGAG
AGCATGAAGTCGGAATCGCTAGTAATCGCGGATCAGCATGTCGCGGTGAATACGTTCCCGGGTCTTGTACACACCGCCC
GTCACACCATGGGAGTGGAATCTGGCAGAAGTAGGTAGCCTAACCGCAAGGAGGGCGCTTACCACGCTGGGTTTCATGA
CTGGGGTGAAGT
敬请关注“灰藻视界”,共筑健康未来!
— 武汉市灰藻生物科技有限公司团队敬上
灰藻生物:我们期待着与客户共同成长,共创生命科学的美好未来!
更新日期:2025-10-25
编制人:小灰
审稿人:小藻